Computel: measuring telomere length and telomeric repeat variant composition from WGS data
![]() Telomere length measurement is key to telomere biology research. Currently available whole genome sequencing datasets contain inherent information about telomere length, which needs to be extracted by bioinformatics tools. This will allow for performing studies on association of telomere length with genomic variations, gene expression, etc.
Telomere length measurement is key to telomere biology research. Currently available whole genome sequencing datasets contain inherent information about telomere length, which needs to be extracted by bioinformatics tools. This will allow for performing studies on association of telomere length with genomic variations, gene expression, etc.
Computel allows for:
- Measuring mean telomere length (MTL) across the chromosomes from whole genome Illumina sequencing data
- Estimating the abundance of non-canonical telomeric repeat variants




Algorithms
Mean telomere length computation
Telomeres have variable length across chromosomes, and across cells. The reads generated via whole genome sequencing (WGS) can be processed to compute the avarage length of telomeres across chromosomes and within a cell population. Computel uses the following steps to estimate MTL:
Step 1. Distinguish telomeric reads from non-telomeric ones
Telomeric reads are those reads that are sequenced from the telomeric regions of the chromatin or from junction of telomeres and subtelomeric regions.
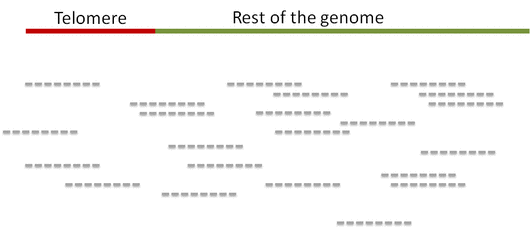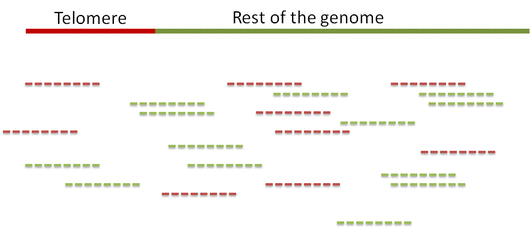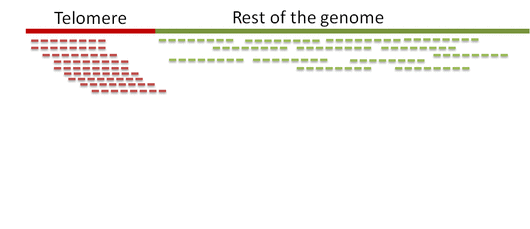
In order to distinguish between the reads Computel first performs step 0.
Step 0. Alignment of reads to a telomeric reference
Unlike equivalent methods for identification of telomeric reads that are based on pattern matching, Computel uses alignment to a specially designed telomeric reference, using Botwie 2. Due to alignment based identification, Computel is on one hand robust against sequencing errors, and sequence variations, and on the other hand it minimizes the number of false positive hits that could come from non-telomeric reads containing telomeric repeats (interstitial telomeric sequences).
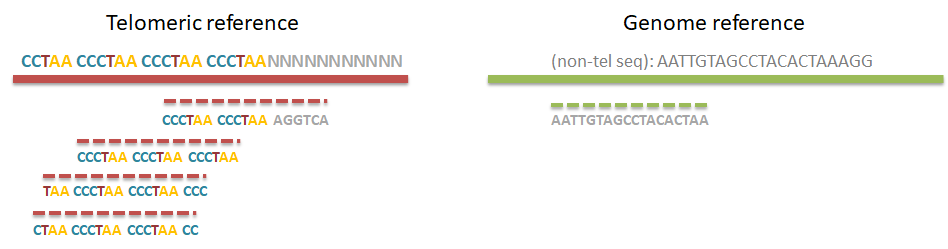
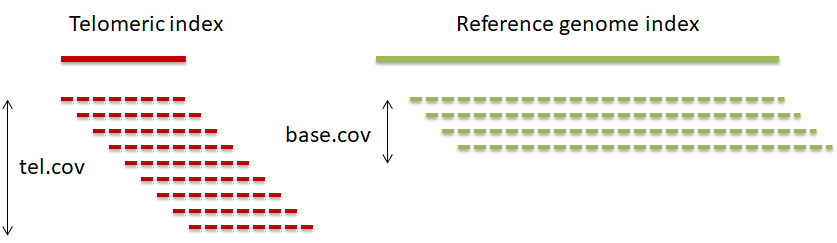
Step 2. Compare the number of telomeric reads with sequencing coverage to compute MTL
The ratio of the coverage at telomeric reference and that at the genomic reference is proportional to the number of telomeric ends and the telomere length and is inversely proportional to read length.
MTL = rel.cov * (rl+pl-1) / (2*n_chr), where: rel.cov = tel.cov / base.cov; tel.cov – mean telomere index coverage; base.cov – mean reference genome coverage; rl – read length; pl – telomeric pattern length; n_chr – number of chromosomes in a haploid genome
Telomeric repeat variant analysis
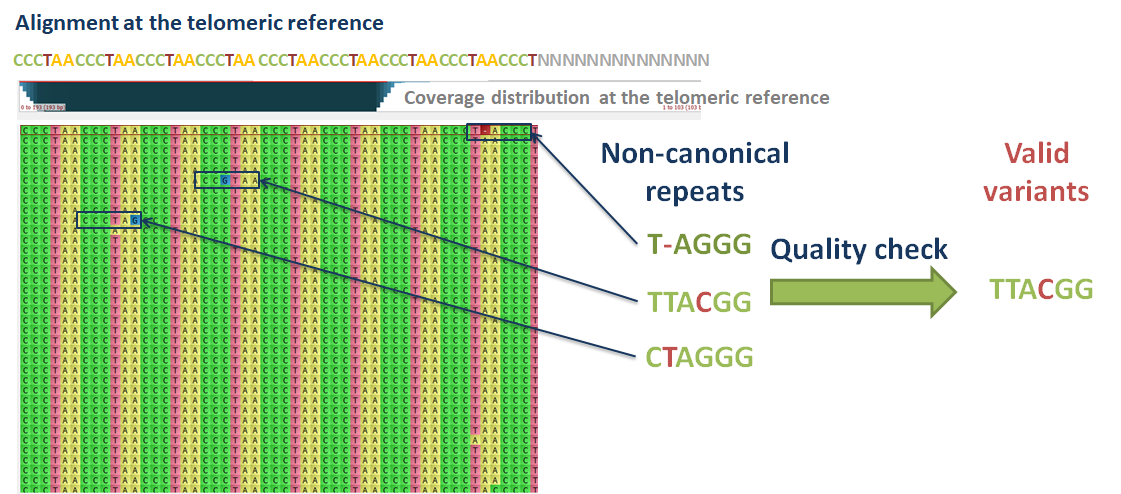
Telomeres do not always possess canonical repeat patterns. For example, in humans, the canonical repeat is ‘TTAGGG’. The telomeres may also contain variants of this pattern, with insertion, deletions and substitutions. The relative abundance and composition of telomeric variants may change during disease development or cellular proliferation. It is thus important to be aware of the distribution of telomeric repeat variants in the studied samples, along with computing their MTL.
Computel uses the telomeric alignment files as input, to estimate abundance of canonical and variant telomeric repeat patterns. The CIGAR strings in the sam files guide Computel for a proper frame to identify repeat variants, and the quality values of the mismatched or inserted bases indicate whether the variant has likely arisen from sequencing errors or from mutations.
Downloads
Computel versions 0.3 and higher work in Linux environment. All the Computel releases are available for download from the github repository.
To clone the current branch, in the terminal type:
git clone https://github.com/lilit-nersisyan/computel.git
Or proceed to Comutel releases and download the release archive.
User manual
Versions of Computel 0.3 and higher have made it quite a straightforward tool to use. To download and get a quick start, read this user manual. A more detailed user manual is available at the Computel github repository.
A quick User Manual
System requirements
You have to have R (version 3.0.3 or higher) and samtools version 1.3 or higher (for Computel v0.4) installed in your system.
Installation
For installation download and uncompress the Computel package in a local directory. The required binaries and files for setup configuration are set in the package. Make computel.sh executable by running ‘chmod +x computel.sh’.
Testing
To test if Computel is set up properly, we advise to run the following command first:
computel.sh -1 src/examples/tel_reads1.fq.gz -2 src/examples/tel_reads2.fq.gz -o mytest
Usage
The basic usage is:
./computel.sh [options] -1 <fq1> -2 <fq2> -o <outputpath>
<fq1> fastq file (the first pair or the only fastq file (for single end reads)
<fq2> fastq file (optional: the second pair of fastq files, if exists)
<fq3> fastq file (optional: the third pair of fastq files, if exists)
<o> output directory (optional: the default is computel_out)
Type ./computel.sh in the terminal for additional options
Output
Two files are generated after successful run:
- tel.length.xls – this file contains information about the provided reads (length, base coverage), organism info (telomere pattern, genome length, number of haploid chromosomes) and the telomere length computed by Computel.
- tel.variants.xls – contains the absolute and relative abundance of the number of telomeric repeat patterns
More options
More details and additional options are described at the Computel github repository.
Citation
Please, use the following citation to read about our software, and cite it in your research:
Nersisyan L, Arakelyan A. Computel: computation of mean telomere length from whole-genome next-generation sequencing data. PLoS One 2015, 10(4):e0125201. PubMed Full text
FAQ
What is the difference between Computel and equivalent tools?
Currently available tools for MTL measurement from WGS do pattern matching to distinguish between telomeric and non-telomeric reads. Computel uses alignment approach. We align the reads to a specially designed telomeric reference. This makes Computel’s algorithm more robust against sequencing errors and telomeric variants arising in different pathological conditions.
What is the unit of measured MTL?
Computel outputs MTL in absalute values, in base pair units.
Can I use Computel for non-human species?
Yes. The default species for Computel is homo sapiens, with default species-specific parameters: telomeric repeat pattern: “TTAGGG” number of haploid chromosomes: 23 genome length: 3244610000 If you’d like to suppl another organism’s WGS data, you should change these parameters with Computel’s options respectively.
Can Computel distinguish between telomeric ends and interstitial telomeric repeats?
Due to the flanking N’s at only one side of the telomeric reference, Computel is able to minimize the number of interstitial telomeric sequences, wrongly classified as telomeric. This makes Computel more specific than other pattern-matchin algorithms.
What's the accuracy of Computel?
Computel is designed to increase its sensitivity by reducing the amount of false negative hits: i.e. unidentified telomeric reads. Compared to pattern-matching algorithms, mismatch mutations and small INDELs do not lead to false negative results, as those still allow for allingment to the telomeric reference. The alignment by bowtie 2 may itself lead to low sensitivity, thus, we have carefully calibrated the alignment options to increase sensitivity (at the same time not affecting specificity).
Computel increases its specificity by reducing the number of genomic reads having telomeric repeats (a.k.a. interstitial telomeric sequences). This is achieved due to the flanking N’s at only one side of the telomeric reference. This makes Computel more specific than other pattern-matchin algorithms.
For tests giving exact values of sensitivity and specificity depending on read length and genomic coverage, please, refer to the supporting information of our paper.
Is Computel limited to certain repeat variants?
No. In comparison with pattern-matching algorithms, Computel checks for any variants present in the telomeric reads. Due to using CIGAR strings generated after the alignment, Computel reduces variant redundancy that could otherwise arise due to frameshift pattern matches (this will be documented in Computel version 2.0 manuscript).
How does Computel distinguish between inherent telomeric repeat variants and sequencing errors?
Computel checks that identified variants pass a certain quality check, based on read qualities provided in the FASTQ files. We have chosen a quality threshold that leads to most accurate results (this will be documented in Computel version 2.0 manuscript).
Should I change the min.seed option?
Min.seed is the seed parameter given to bowtie 2 for alignment. The more the min.seed, the more the specificity and less the sensitivity of Computel. We have performed multiple tests on reads ranging between 20-150 bp in length, and found that min.seed of default value 12 is ideal. We don’t recommend changing this value, unless you have a clear reason for doing so.
Does Computel account for aneuploidy?
Computel does not explicitly account for aneuploidy or any other changes in the number of telomeric ends or extra-chromosomal telomeric sequences. However, if you are aware of the changes in the number of chromosomes in your samples, you can change the number of haploid chromosomes in the Computel’s input and this will let you adjust the mean telomere length value you obtain.
Discussion forum
If you’d like to post a question or view users’ discussions, please, visit the Computel Google Group.