The Telomere Maintenance Mechanisms (TMM) Project
There are t![]() wo mechanisms that maintain the length of telomeres in healthy and cancerous tissues: telomerase-dependent and alternative (ALT) mechanisms. This project is aimed at fostering research on telomere maintenance mechanisms (TMM).
wo mechanisms that maintain the length of telomeres in healthy and cancerous tissues: telomerase-dependent and alternative (ALT) mechanisms. This project is aimed at fostering research on telomere maintenance mechanisms (TMM).
- We provide Telomerase and ALT TMM pathways that we’ve curated based on literature and gene expression data.
- We’ve created a Cytoscape app for assessment of TMM activity from gene expression data.
-
- You may use these pathways and our app to assess which TMM mechanism is active in your samples, if you’ve got gene expression data for them.
- Most importantly, we believe that many researchers may contribute their knowledge or data to improve these pathways and to deepen current understanding of molecular mechanisms leading to TMM activation.




About
The mechanisms employed by highly proliferative cells to maintain the length of their telomeres may vary, but are classified into telomerase dependent and independent mechanisms. Activation of a particular telomere maintenance mechanism (TMM) specifies the behavior of a cell. In cancer cells, the TMM activity is a predictor of tumor aggressiveness and cell survival.
It also predicts the cell’s response to anti-cancer therapies targeting TMMs. Therefore, it is important to understand the factors leading to activation of TMMs. While there is a considerable amount of experimental data on TMM, combination of data- and knowledge-driven approaches based on utilization of high-throughput gene expression have the potential to foster further developments in this direction.
We have reconstructed in silico models for TMM phenotype prediction. These models represent pathways that include the key proteins, RNAs, and processes involved in the TMMs and the functional interactions between them. These models have been tested on experimentally annotated samples and shown to be valid predictors of TMM activity.
How to use the TMM pathways?
1. Download the TMM app for Cytoscape.
2. Download TMM pathways of desired version (last version recommended).
TMM pathways version 0.0
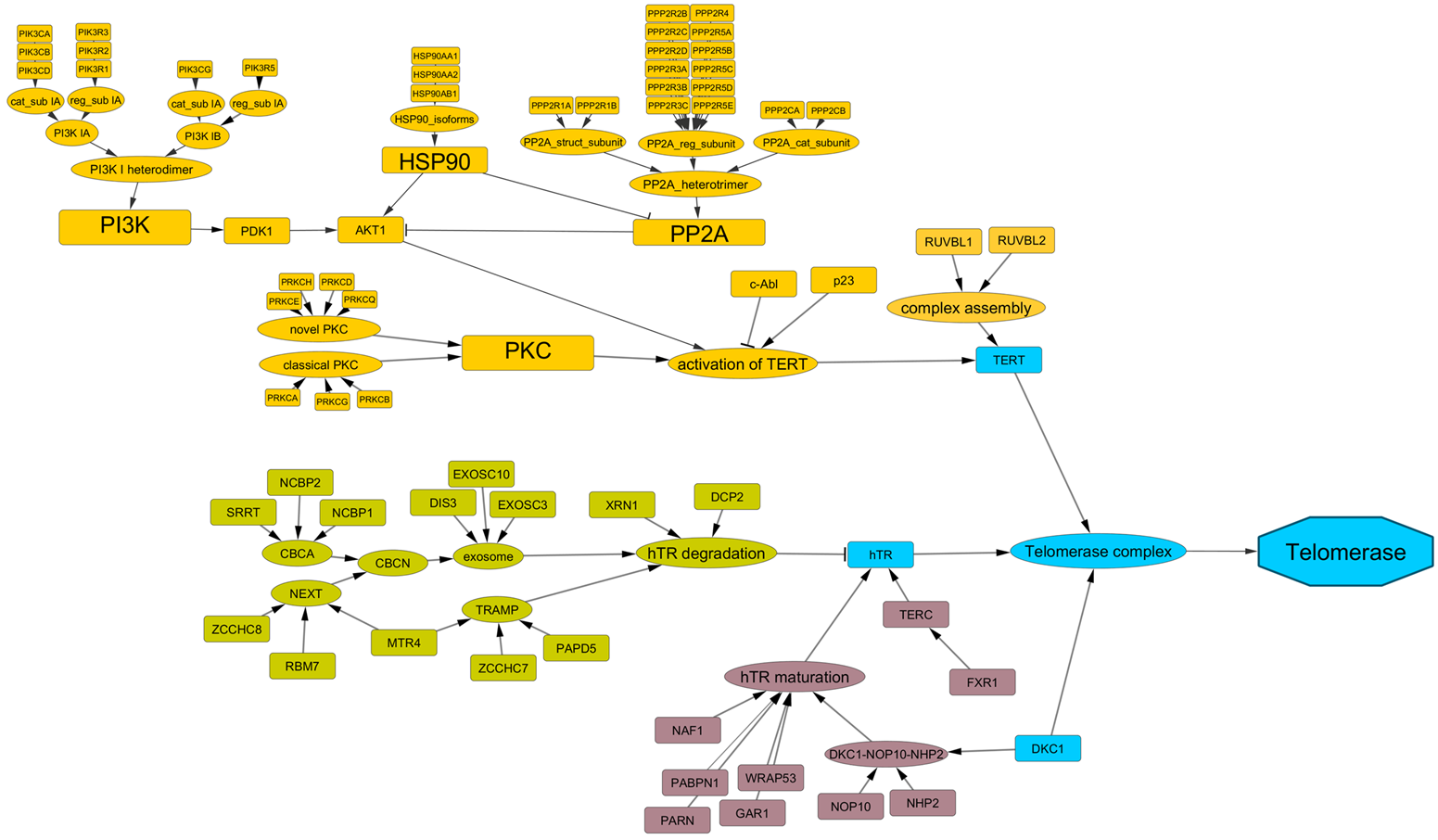 ALT pathway
ALT pathway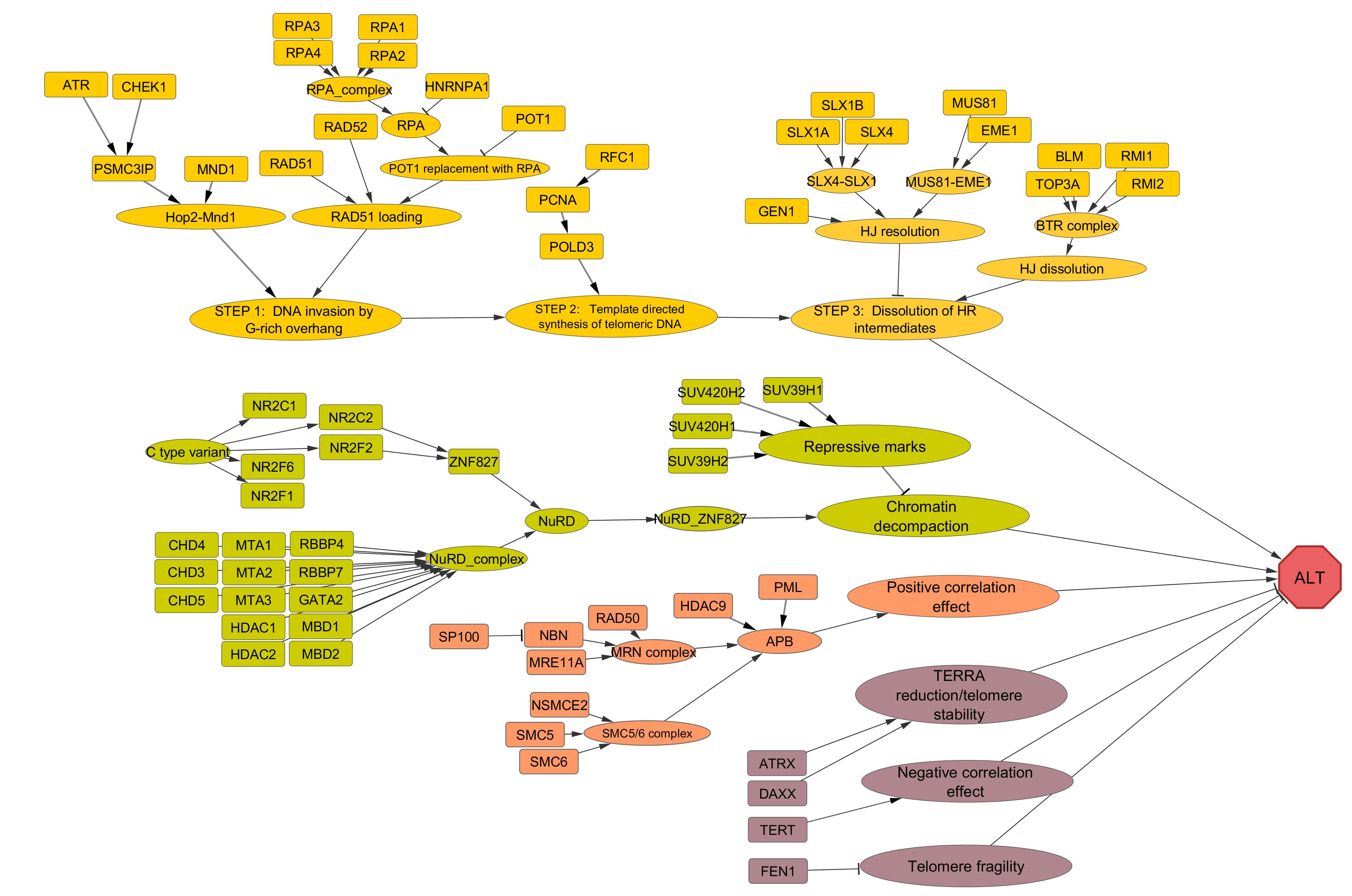
TMM pathways version 1.0
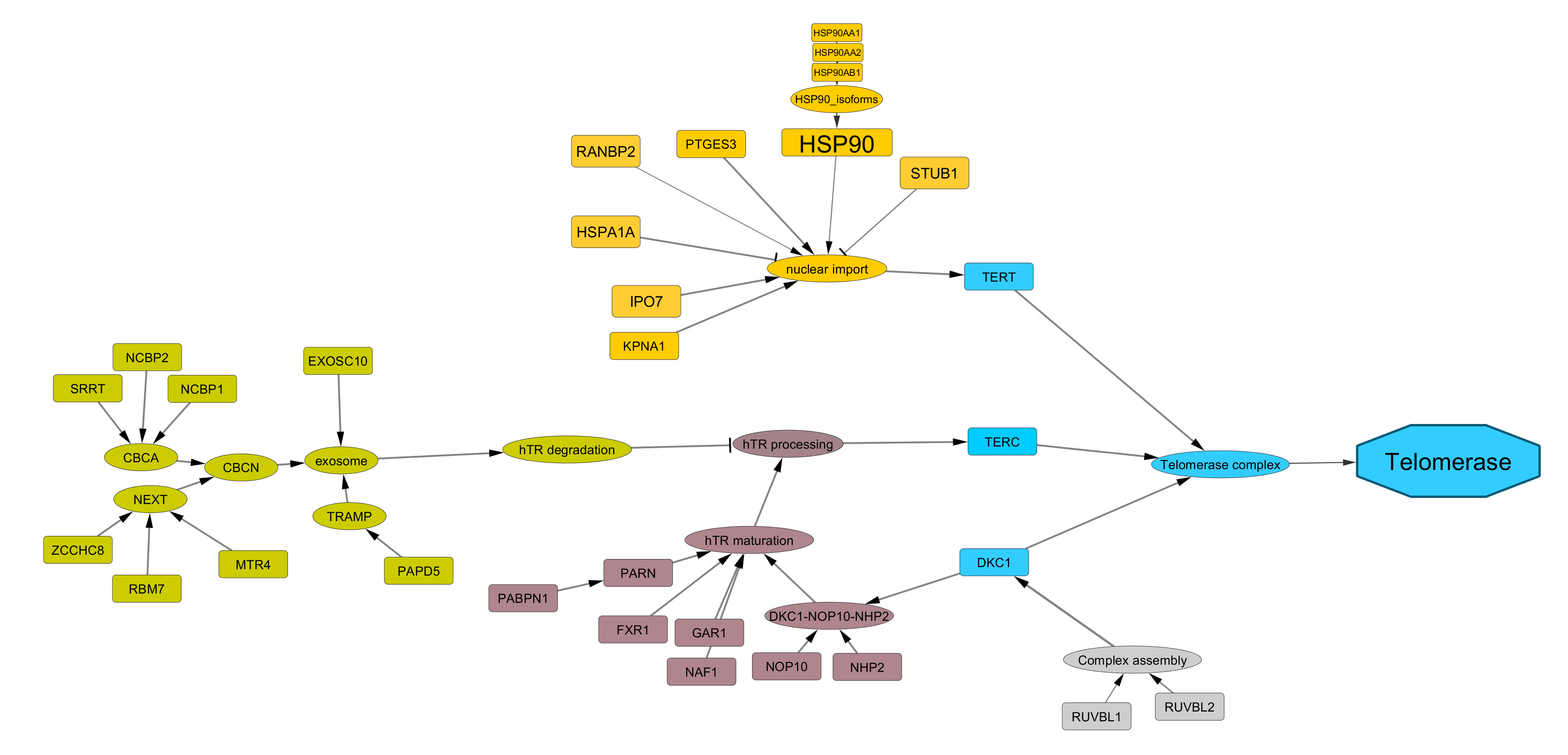
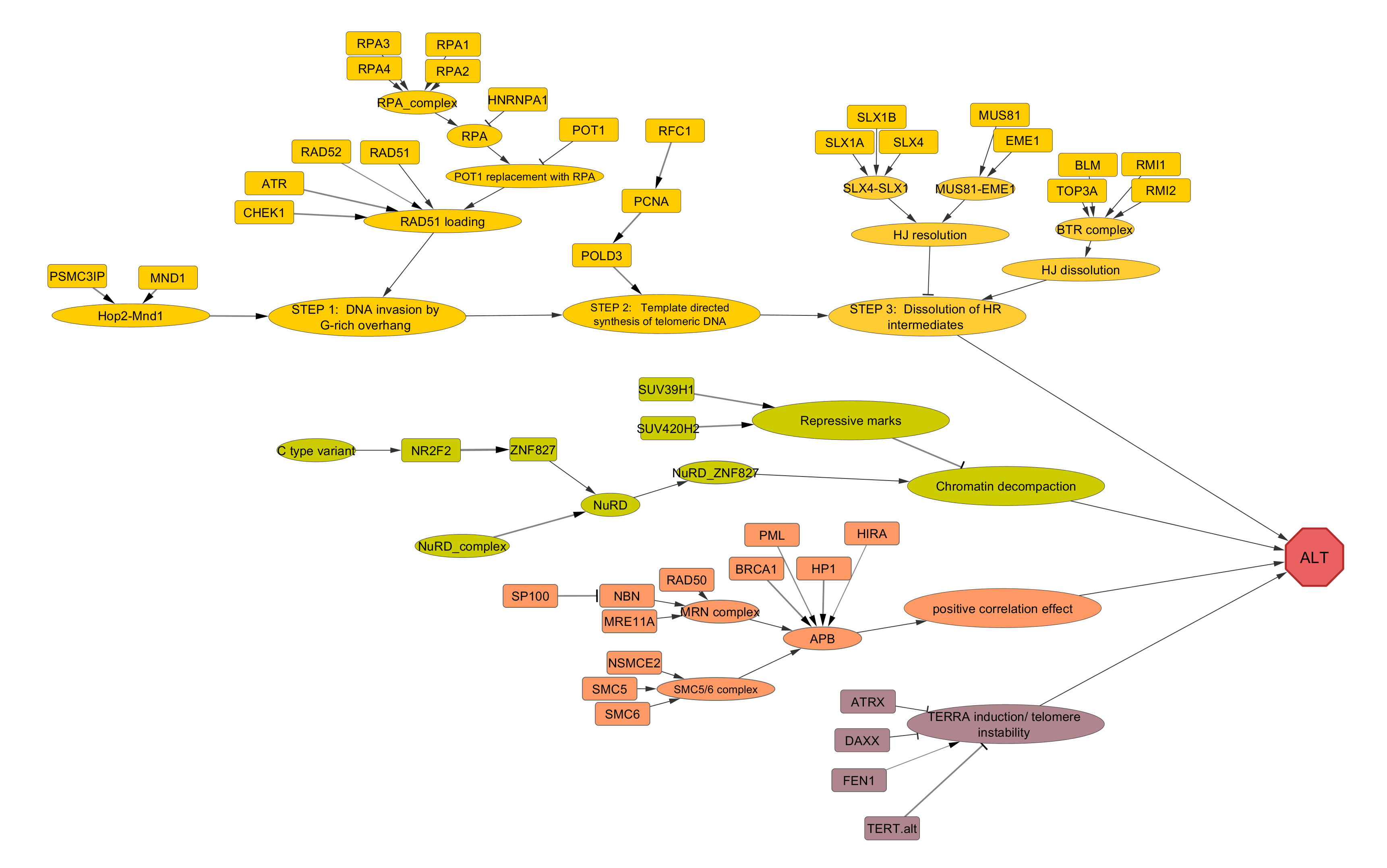
TMM pathways version 2.0
TEL (Telomerase-dependent) pathway
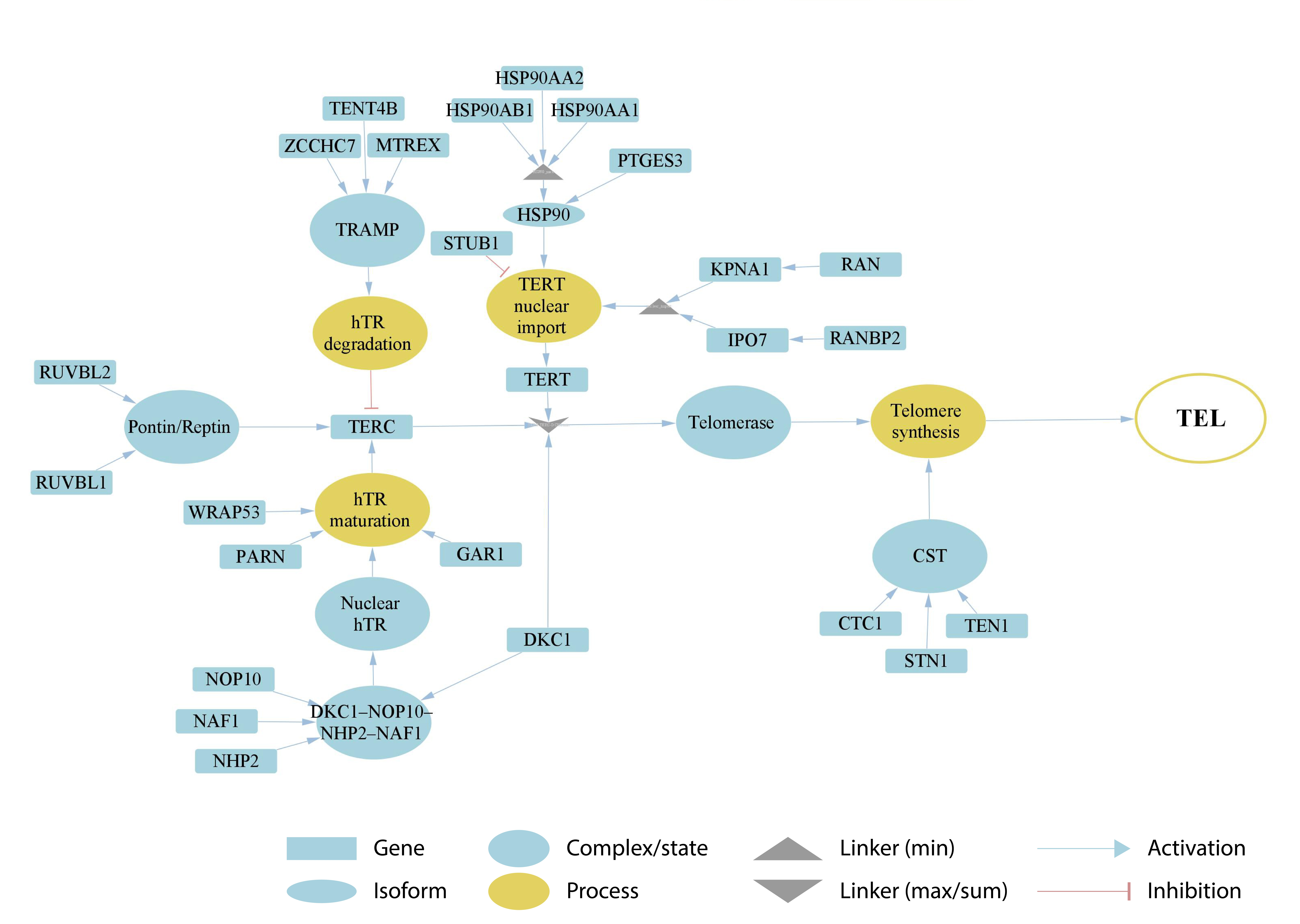
ALT (Alternative lengthening of telomeres) pathway
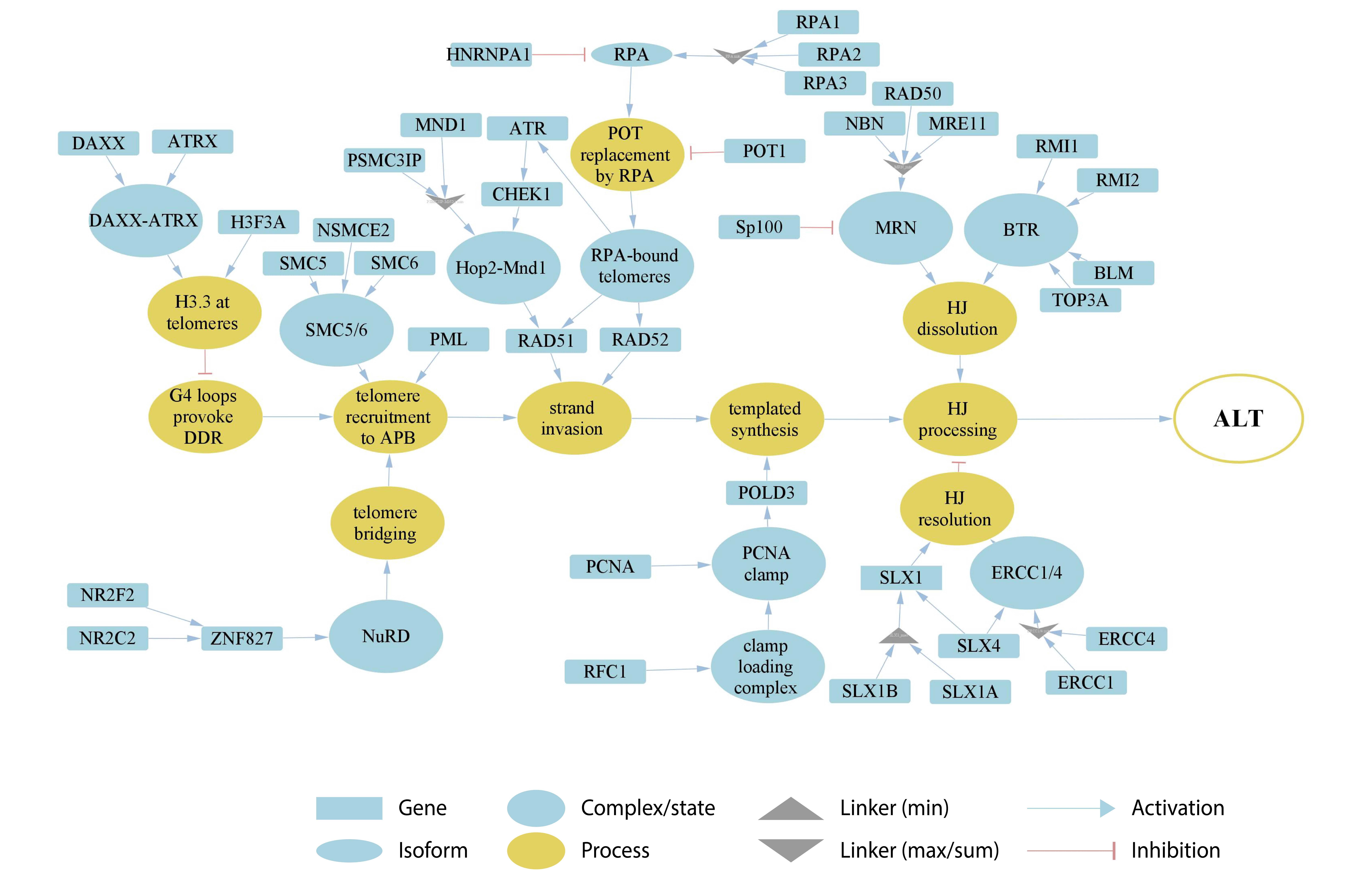
3. Supply gene expression data of your samples.
4. Use the TMM app to evaluate the activity of the two TMM pathways.
5. Assess the TMM states of your samples
Supply gene expression data to the TMM app, and see the distribution of your samples in the TMM space: from low-to-high ALT, and low-to-high telomerase activity.
6. Contribute your knowledge or data to improve the TMM pathways
If you possess experimental annotations of TMM phenotypes for your gene expression datasets, you may use these to validate the TMM assessment results of the TMM app. Based on your data, you may suggest corrections for the pathways and submit the corrected versions, as well as make your data available for other researchers.
Downloads
TMM app
Downlaod TMM app for Cytoscape:
Dependencies
- You have to have Cytoscape installed on your system:
- You have to have the PSFC app version 1.1.6. If you don’t have it, Cytoscape will install it automatically during TMM installation. The link to PSFC app is here:
Download PSFC app for Cytoscape
Networks and examples
Networks
ALT – Telomerase TMM pathways
Examples
Example files are to help you get started with the TMM app. Each archive contains the full set of input files, as well as the output files that should be generated.
Each example zip archive contains a network file, a gene expression data file, a TMM labels’ file and a report folder, where the .pdf and .xls outputs generated by TMM are present.
Example #4. Network: tmm_net_v2.0.xgmml, Data: data#1: Cell lines.
Example #3. Network: tmm_net_v2.0.xgmml, Data: data#2: Liposarcoma.
Interactive networks will be uploaded in the future: where you click on the edge and it gives the reference of the current interaction. To this point, detailed information on how the pathways are curated is available in the citation.
TMM genes
To see the list of those genes that are either included in TMM pathways, or tested but not included, and those to be tested, click this link.
TMM User Guide
Installation
TMM has been tested to work with Cytoscape version 3.4.0 and higher on 64 bit systems.
Dependencies
Install Cytoscape
The TMM app works with Cytoscape. If you don’t have it on your system proceed to Cytoscape website and follow installation instructions.
Install TMM 0.5
Go to Apps -> App Manager -> choose TMM from the list of available apps and click on the install button.
TMM 0.5 works with PSFC 1.1.6 Cytoscape app as its dependency. If you don’t have it, Cytoscape will install it automatically during TMM installation. In case of successful installation, the “TMM” app tab should appear in the west panel of Cytoscape.
User flow
Load the network
To load a network into Cytoscape,
Go to File -> Import -> Network -> File -> select the TMM network you’d like to load -> click Open
Download the TMM network:
Literature and data curated TMM networks are available here.
Supply input data
In the TMM tab, the first panel is for supplying input files and directories to TMM.
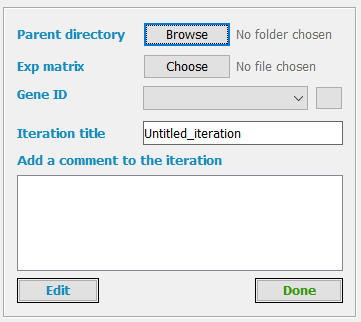
Parent directory
The parent directory is the folder where the iteration directory will be created by TMM.
Click Browse -> in the new window select an existing directory -> click Open
Expression matrix
The expression matrix file contains gene expression data in a tab delimited format:

The column GeneIDs should contain gene identifiers that are also available in the Node Table of the Network loaded in Cytoscape. The networks downloaded from the TMM webpage identify genes by their Entrez IDs (the “entrez” column in the network node table). Thus, in case of using those networks the user should have Entrez IDs in the GeneIDs column.
ExpG1S1 is the expression value for Gene 1 in sample 1.
To load the expression matrix file:
Click Choose -> in the new window select the expression matrix file -> click Open
Example gene expression files are available for download from Downloads.
Gene ID
The Gene ID specifies which column in the Cytoscape Network Node Table corresponds to gene identifiers also present in the expression matrix file. The networks downloaded from the TMM webpage identify genes by their Entrez IDs. Therefore, the user should choose the “entrez” column. The user will be notified In case if no node from the selected column is found in the first column of the gene expression matrix file.
Iteration title
The pathways may be modified by adding nodes, changing the locations of the nodes in the pathway, and deleting nodes. We name one such change an iteration. You may try different iterations to see which one performs the best in terms of prediction accuracy. The TMM app will create a separate directory for each iteration with the supplied iteration name, and will export all the files to that directory.
Add a comment to the iteration
You may add a description to the current iteration for further reference (e.g. in this iteration, the node FEN1 was removed). The comments will appear in the final report file.
Done and Edit
After submitting all the input files, click the “Done” button. The TMM app will create a directory for the iteration, will export the network file, and will create a file containing fold change values from the supplied expression matrix file. If any of the inputs is not valid, TMM will notify you. If all the files are valid, the input buttons will get disabled, and the rest of the TMM tab functionality will be enabled. To modify the input the you may click the “Edit” button.
Indicate TMM annotation labels
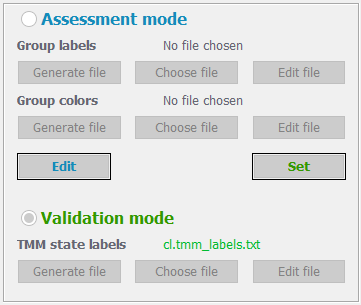
If the samples have been experimentally tested to detect either ALT or Telomerase activity you may supply the results to validate the pathways. In this case, press the “Validation mode” radiobutton, otherwise choose the “Assessment mode” option to simply assess the relative activity of ALT and Telomerase pathways in your samples.
In case of selecting the Assessment mode option, the user may supply additional information to color the samples according to custom grouping. For this, press the “Generate file” button under the “Group labels” text. You will be asked to specify the path and name of the file – please, supply the full name with the extension, e.g. “my_group_labels.txt”. The app will add a correctly formatted content to the specified file, where the first column contains the names of your samples, and the second column indicates the group label of each sample. The app will assign all the samples to the group label “unknown”. You may edit the file in place, or edit it in your system. In case you already have a group labels’ file, you may supply it by pressing the “Choose file” button.
The group colors’ file contains two columns: the first one with group labels (should coincide with those specified in the group labels’ file) and the second one with colors (hex codes in the format #000000) for each group label. The “Generate file” button under the “Group colors” label will again ask you to specify the path and name of the file – please, supply the full name with the extension, e.g. “my_group_colors.txt”. After that the app will place a correctly formatted content for you – based on the supplied Group labels’ file. You may then choose to edit it. Again, you can simply provide the file if you already have it by clicking the “Choose file” button.
After supplying the group labels and colors, click “Set”. You can return and edit these anytime with the “Edit” button. The group labels and colors will be used in the PDF report generated by TMM.
In case of selecting the Validation mode option, you should supply the experimentally annotated TMM labels. You may click the “Generate file” button, which will create a text file in the correct format, where, however, all the samples will have ALT-/Teloemerase- annotations. If your samples are ALT positive, you should change the “-“ sign with “+” under the ALT column. If your samples are Telomerase positive, then those should have “-“ replaced with “+” under the Telomerase column. The file is in the following tab delimited format:

If you already have the TMM labels file, you may specify it by hitting the “Choose file” button. If you’d like to modify the newly generated or selected file, you may click the “Edit file” button, which will open the file with your system’s default text file editor (usually Notepad).
Example data with TMM labels is available for download from Downloads.
Save settings
The supplied input files, folder names, comments, as well as the TMM annotation options may be saved by clicking the “Save settings” button, and will be reloaded next time the TMM app is launched.
Run TMM
After all the inputs are supplied, TMM does the following steps in order:
Add/update fold change values
Imports the fold change values of genes into the Network Node Table in Cytoscape. The fold change values are computed from the gene expression data. For each gene, the mean expression value is computed across the samples, and each expression value is then divided by this mean value. If the mean is 0 (in case all the samples are zero), the fold change for this gene is assigned a value of 1 for all the samples. Fold changes of 1 are also assigned in case of NA’s. The generated fold change values are stored in the iteration directory with the name “fc_iterationTitle.txt”. When clicking the “Add/update fold change values” button, these fold changes are imported into Cytoscape and can be seen in the Node Table with separate columns for each sample.
Run PSF
Performs a call to PSFC (Pathway Signal Flow Calculator) app to compute pathway PSF activity values of ALT and Telomerase pathways. The PSF algorithm computes the signal propagation from pathway inputs to the outputs (ALT and Telomerase nodes, in our case) based on fold change values and the topology of the pathway. Shortly, for each source -> target interaction, it multiplies the fold change of source to the target and assings the product to the target, if the interaction is of “activation” type. The inverse (1/SourceFC) of source fold change is multiplied by the fold change of the target node if the interaction is of type “inhibition”. The interactions of type “activation” have a delta shaped edge target in the network, while the “inhibition” type has a T-shaped edge target. If the target node has multiple incoming edges, the edge signals are usually summed. Some nodes, however, represent complexes, where presence of all the subunits is necessary for it to function. In this case the minimum value of incoming signals is taken. If the at least one component is enough for the target node to function, the maximum of all the incoming signals is assigned to the target node. Please, see the citation for more details.
Click the “Run PSF” button to perform a call to PSFC. The PSF activity values at each node will be computed and kept in summary files in the iteration directory, as well as will be imported to the node table. Of interest are the PSF values at the “ALT” and “Telomerase” nodes, since they reflect the activity of the whole pathways. PSFC also computes significance p values for PSFs by bootstrapping. You may increase the number of bootstrap cycles to increase the power, or you may decrease the number of bootstrap cycles to make the computations faster, but at the expense of having less statistical power. The number of bootstrap cycles can be set in the “Bootstrap” textfield.
Generate report
Generates a report describing the pathway activity statistics. TMM generated two types of reports in a “report” folder residing in the iteration directory: the PDF file containing statistics and charts, and the “.xls” formatted file containing PSF values of ALT and Telomerase pathways for all the samples. The first page of the PDF report contains the input options, the iteration comment and the obtained statistics. The other pages contain volcano plots and boxplots (if TMM labels are supplied) for ALT and Telomerase pathways, and a 2D-plot where the Telomerase the ALT pathway PSF scores are on the x and y axis, respectively. If TMM labels were supplied, the TMM app also computes the SVM-based thresholds separating ALT from non-ALT and Telomerase from non-Telomerase samples and draws horizontal and vertical lines on the 2D plot. To open the report file, click the “Open” button.
Run all
All of the three steps above may be run with one click to the button “Run all at once”.
Visualize PSF results

The PSF values are visualized in Cytoscape with node colors on a red-white-blue scale, where red and blue represent high and low PSF values, and white represents PSF values equal to 1. The sample is chosen with the drop-down menu, and visualized by clicking the “Viz” button.
FAQ
To be added.
Citation
- Nersisyan L, Arakelyan A. A transcriptome and literature guided algorithm for reconstruction of pathways to assess activity of telomere maintenance mechanisms. bioRxiv 200535. doi: https://doi.org/10.1101/200535
- Please, cite the following PhD thesis, until the paper is out.
Submissions
You’ve got an evidence that some of the nodes and edges in the available TMM networks are not accurate?
You’ve got an evidence of new players not yet available in the TMM networks?
You have novel gene expression data from experimentally TMM annotated samples?
You’ve tested your data to validate the TMM networks?
Share your knowledge and data! Let us improve our understanding of telomere maintenance mechanisms by submitting your networks and data.
